Nextflow
Nextflow is a relatively new vertical execution stack for bioinformatics and takes a unique approach by providing a groovy based DSL that is reactive streams based. Nextflow supports multiple backends including Docker and supports any POSIX compatible system. Windows systems are only supported through WSL. Below outlines how to run Nextflow locally using Docker.
Prerequisites
Getting Started
The following software and workflow are required and will be downloaded, installed, and used as part of this tutorial:
Download the worked examples repository by one of the following methods.
- Clone with Git
1git clone https://github.com/DNAstack/dnastack-client-library-worked-examples.git - Or download and unzip
It is assumed that all commands will be run from the root directory of this repository on your local machine. From the directory where you’ve downloaded the examples repository.
| |
Download and install Nextflow by one of the following methods.
- Using cURL or direct from this link, and allow execution permission
1 2 3curl -o nextflow -L https://get.nextflow.io # Or directly from web link chmod +x nextflow - Or using Conda
1conda install -c bioconda nextflow
Running the workflow
To run the workflow:
| |
Output files can be found in the current directory.
Nextflow Tower
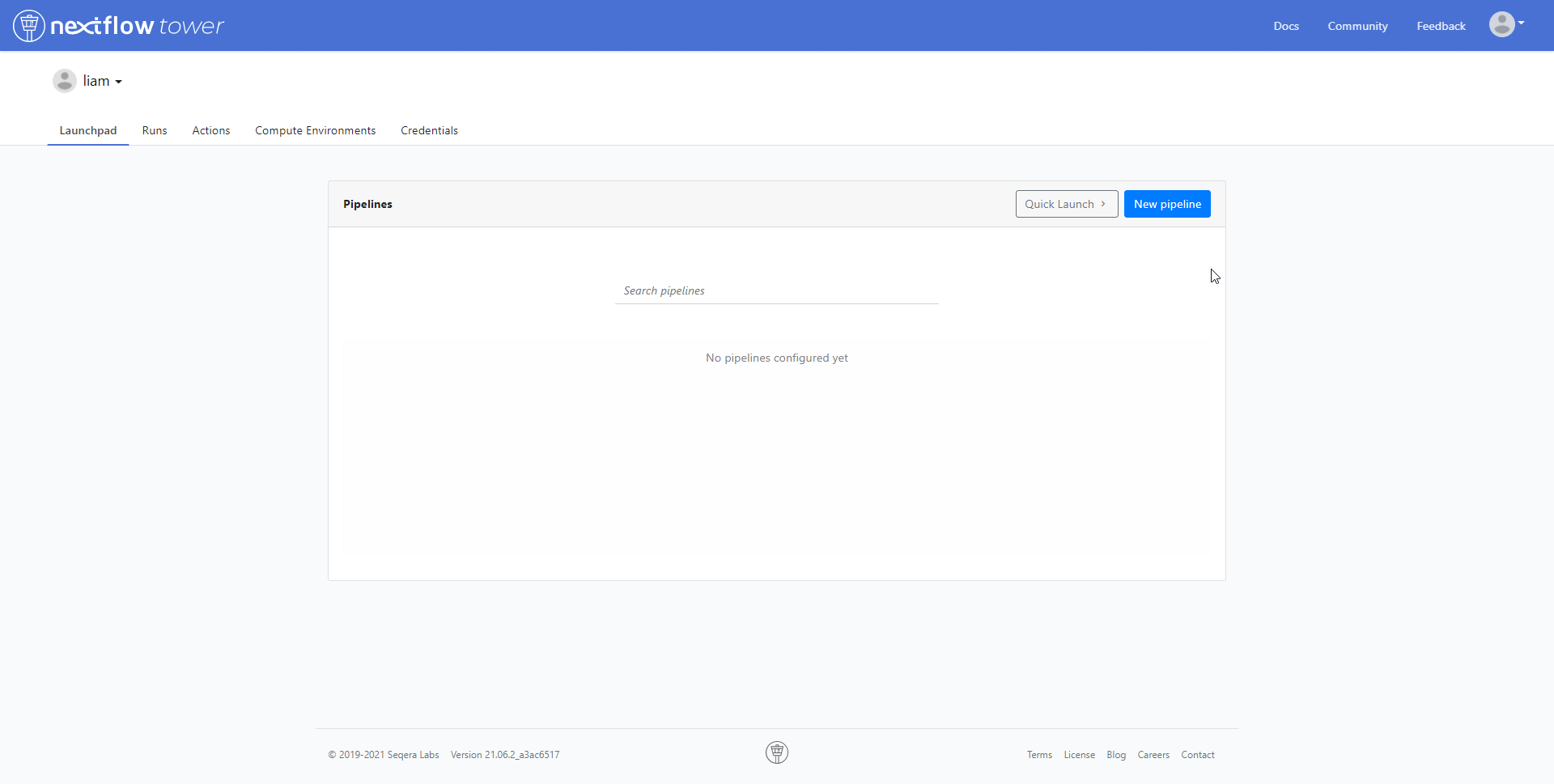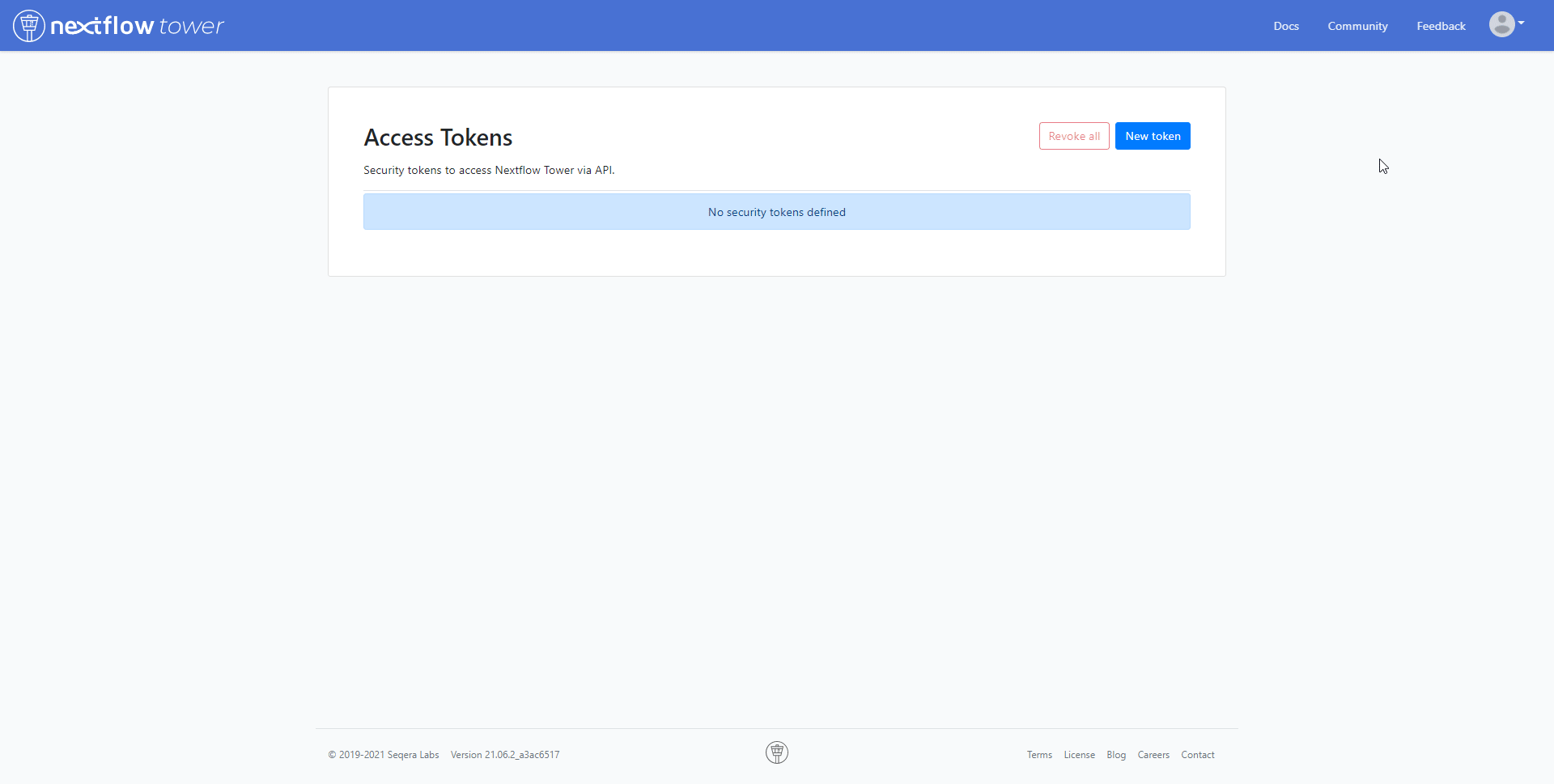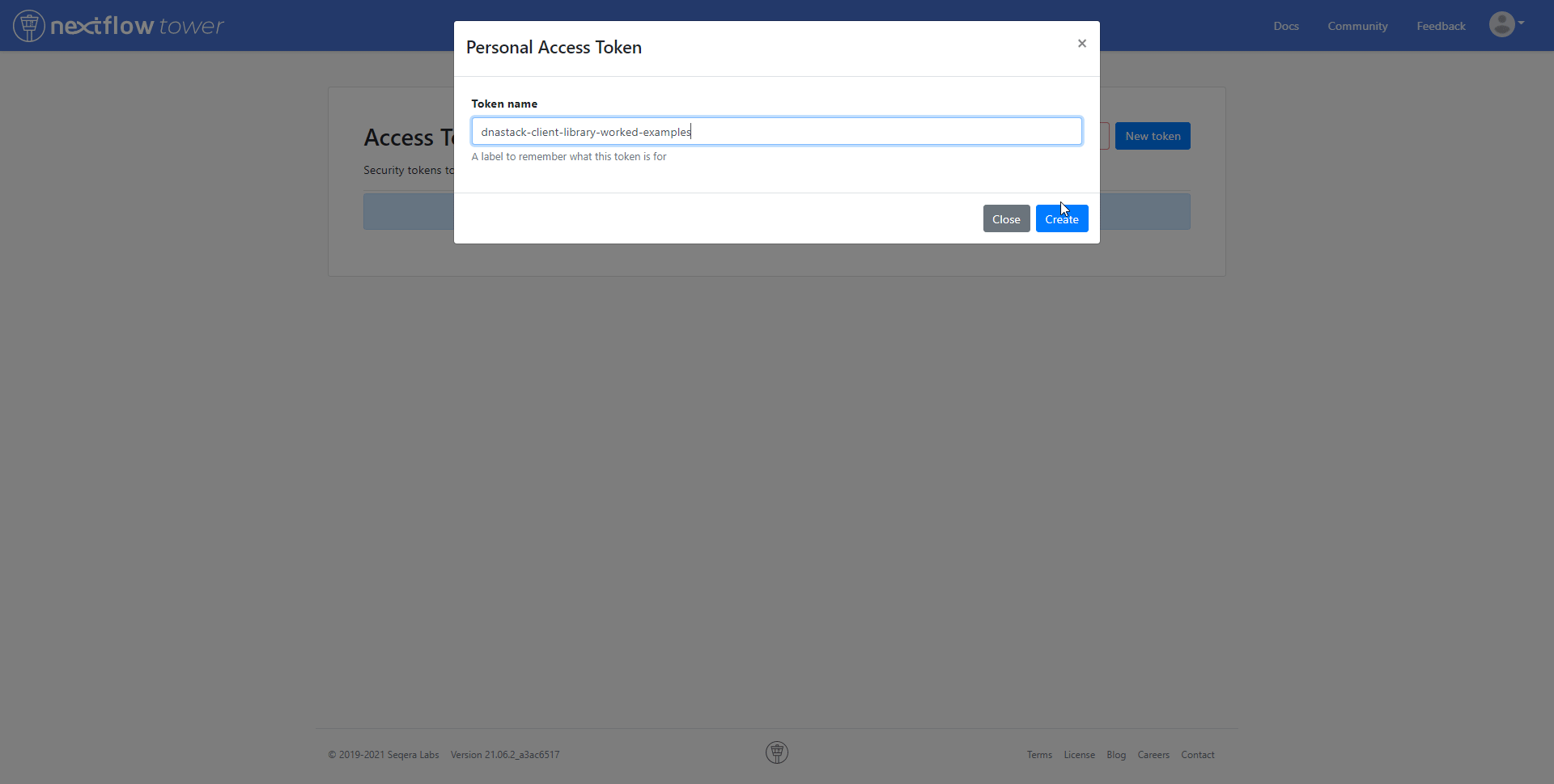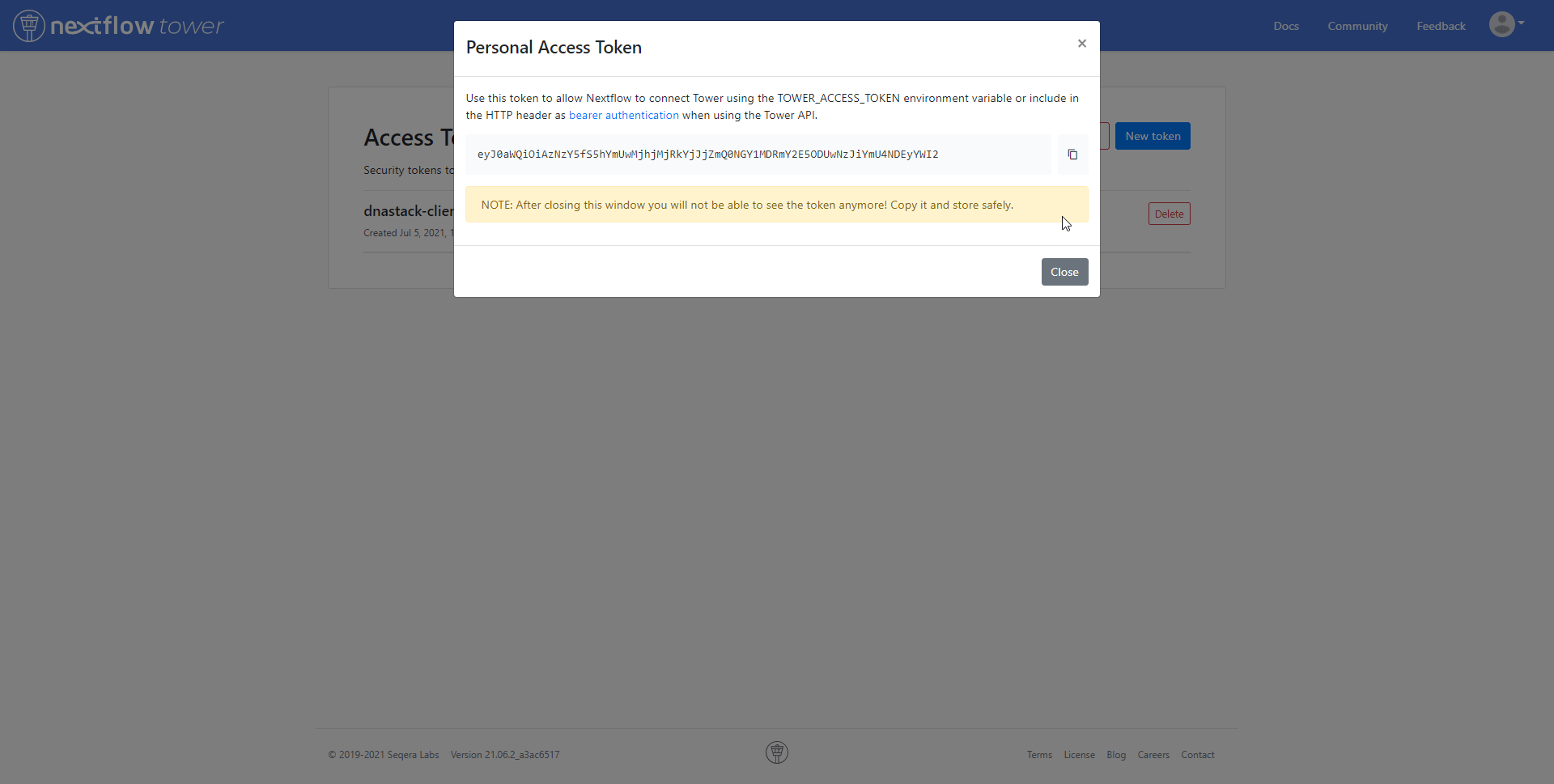
Optionally, Nextflow Tower allows remote monitoring, logging, and observability to Nextflow workflows through a centralized command-post.
To use Netflow Tower with the workflow, add the following to the configuration file nextflow.config. Replace <YOUR TOKEN> with your specific access token.
| |
Your access token can be found by navigating to the user menu, and to Your tokens.
Details about the workflow job can be found by navigating to runs.